ERBKRANKHEIT
CF entsteht durch Veränderungen in den Chromosomen und wird von Eltern an ihre Kinder vererbt.
4% der Schweizer Bevölkerung sind CF-Träger
Die Cystische Fibrose entsteht durch einen Fehler im Erbgut, den die Eltern an die Kinder weitergeben. Das menschliche Erbgut ist auf 23 Chromosomen verteilt, welche jeweils doppelt angelegt sind. Falls eines dieser Chromosomen einen Fehler enthält, kann sein unveränderter „Zwilling“ diesen ausgleichen. Diese Situation trifft auf die Eltern von CF-Betroffenen zu. Sie sind nicht krank, nur Träger einer Veränderung auf dem Chromosom 7 – meist ohne es zu wissen. In der Schweiz sind rund 300 000 Personen Träger einer solchen CF-Mutation.
4% der Schweizer Bevölkerung sind CF-Träger
Ein Kind erhält jeweils ein Chromosom 7 des Vaters und eines der Mutter. Wenn ein Kind von zwei CF-Trägereltern das jeweils veränderte Chromosom erhält und somit kein gesundes Chromosom den Fehler ausgleichen kann, treten die CF-Symptome auf. Die Wahrscheinlichkeit, dass dieser Fall bei einer Schwangerschaft eintritt, ist 25%.
Detaillierte Information zum Krankheitsbild
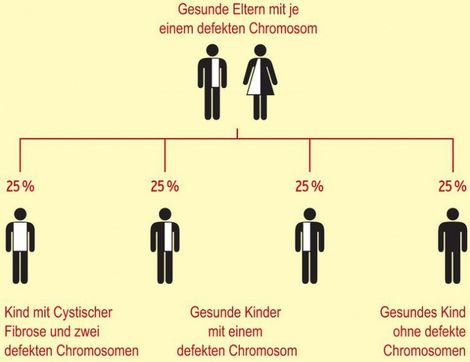
Vererbung bei CF
Quelle:CFCH
DAS KRANKHEITSBILD IM DETAIL
Wer ist betroffen?
[04.2014] Die Cystische Fibrose (CF) wird autosomal-rezessiv vererbt. 4% der Bevölkerung sind Erbträger. Sind beide Partner Erbträger, ist bei 4 Kindern statistisch 1 Kind von CF betroffen, 2 Kinder sind Erbträger und 1 Kind ist weder Erbträger noch krank. Ein Kind kann also nur dann CF haben, wenn beide Elternteile Merkmalsträger sind.
Häufig wird die Cystische Fibrose erst später diagnostiziert. Die Krankheit ist bei verschiedenen Patienten unterschiedlich stark ausgeprägt, und auch der Zeitpunkt, an dem die Krankheit erkennbare Symptome zeigtund dann diagnostiziert werden kann, ist unterschiedlich. Die Krankheit trifft sowohl Knaben als Mädchen
Definition und Genetik
Die Cystische Fibrose ist eine der häufigsten Erbkrankheiten. Sie tritt vor allem in der weissen Bevölkerung auf. Ungefähr 1 auf 2500 Neugeborene, Mädchen gleich häufig wie Knaben, sind betroffen.
Erbkrankheiten sind Krankheiten, welche durch Veränderungen in den Chromosomen entstehen und welchevon den Eltern an ihre Kinder weitervererbt werden. Das menschliche Erbgut ist auf 46 Chromosomen verteilt. Die Chromosomen 1 bis 22 sind jeweils doppelt angelegt. Dazu kommen 2 sogenannte Geschlechtschromosomen, 2 X-Chromosomen bei Frauen, 1 X- und 1 Y-Chromosom für die Männer. Die Erbinformation ist also für die Chromosomen 1 bis 22 jeweils doppelt angelegt. Wenn ein Gen auf einem Chromosom verändert ist, also eine sogenannte Mutation aufweist, und deshalb seine Funktion nicht ausüben kann, so kann das Gen auf dem anderen, gleichen Chromosom die Funktion übernehmen. Diese Situation trifft auf die Eltern von CF-Betroffenen zu, welche ein verändertes CF-Gen und ein zweites, unverändertes Gen haben. Sie sind nicht krank und tragen diese Mutation ohne es zu wissen. In der Bevölkerung ist zirka jeder zwanzigste bis fünfundzwanzigste Träger einer solchen Mutation (d.h. 4-5 von 100). Kinder erhalten jeweils ein Gen des Vaters und eines der Mutter. Wenn nun zufälligerweise ein Kind von zwei CF-Trägereltern das veränderte Gen erhält (in 25% der Kinder dieses Ehepaares) und somit kein gesundes Gen die Funktion erfüllen kann, zeigt es Symptome der Cystischen Fibrose.
Die Mutation der CF kennt man seit 1989. Das Gen befindet sich auf dem Chromosom 7. Es können viele verschiedene Mutationen im Gen auftreten, welche zu den Symptomen der CF führen. Die Mutationen sind
alle durch Buchstaben und Zahlen gekennzeichnet. Bis jetzt wurden ca. 1900 Mutationen im CF-Gen gefunden. Die häufigste Mutation trägt den Namen delta F508. Es gibt auch eine Mutation, welche nur in der Schweiz oder bei Schweizer Auswanderern gefunden wird, sie trägt den Namen 3905 insT.
Was für Folgen hat diese CF-Mutation
Unser ganzer Körper ist aus Zellen aufgebaut. Diese Zellen haben eine Zellwand und ein Zellinneres. Die Zellen stehen mit ihrer Umgebung in ständigem Kontakt und Austausch. Der Austausch und Kontakt geschieht über spezialisierte Strukturen (Eiweisse) in der Zellwand. Die Zellen von CF-Betroffenen haben einen Funktionsdefekt eines dieser Eiweisse in der Zellwand (es heisst cystic fibrosis transmembrane regulator, abgekürzt CFTR). Dadurch ist der Austausch von Salzen vom Zellinneren zur Umgebung und zurück gestört. Besonders folgenschwer wirkt sich dieser Funktionsdefekt überall dort aus, wo Flüssigkeiten im Körper gebildet werden, in der Lunge, der Nase und den Nasennebenhöhlen, der Bauchspeicheldrüse, der Leber, dem gesamten Verdauungstrakt, den Schweissdrüsen und den Fortpflanzungsorganen. Zellen, welche die Aufgabe haben, Schleim oder eine Flüssigkeit zu bilden, produzieren allzu zähe und dickflüssige Flüssigkeiten. Die Folgen für die Funktion der verschiedenen Organe wird weiter unten erklärt.
Diagnose
Seit dem Jahr 2011 werden alle Neugeborenen in der Schweiz auf CF untersucht (Neugeborenen Screening durch Fersenbluttest). Mit dem Schweisstest steht zudem eine einfache und schmerzfreie Untersuchung zur Diagnose der CF zur Verfügung, weil der Funktionsdefekt des Zellwandeiweisses (siehe oben) auch in den Schweissdrüsenzellen wirkt. Für diesen Test werden zuerst die Schweissdrüsen zur Schweissproduktion angeregt, anschliessend wird mittels kleinen Fliessblättern etwas Schweiss aufgesaugt und die Salzkonzentration gemessen. Ist diese zu hoch, ist dies verdächtig für eine CF.
Ist die Salzkonzentration im Schweiss zu hoch, wird in den Chromosomen des Patienten nach Mutationen gesucht. Werden dort auf beiden Chromosomen Nr. 7 Mutationen im CF-Gen gefunden, so ist dies beweisend für das Vorliegen einer Cystischen Fibrose. Diese Laboruntersuchung kann, wenn eine seltene Mutation vorhanden ist, mehrere Wochen in Anspruch nehmen.
Folgen in den Atemwegen
Die Auswirkungen auf die Atemwege stehen meist im Vordergrund und bestimmen den Schweregrad der Krankheit. Normalerweise bilden schleimproduzierende Zellen in der Bronchialschleimhaut eine feine Schleimschicht, welche mit den Flimmerhärchen auf der Zelloberfläche zum Mund transportiert wird und die eingeatmeten Schmutzteilchen und Bakterien wieder aus der Lunge abtransportiert. So reinigen sich die Lungen stets selbst. Bei CF-Betroffenen wird von den schleimproduzierenden Zellen ein abnorm zäher, klebriger Schleim gebildet. Dieser kann nur schlecht oder gar nicht abtransportiert werden und bleibt liegen. Die feinen und feinsten Bronchien werden dadurch verstopft und können keine Luft mehr transportieren, die Lunge wird nur unvollständig mit Luft gefüllt. Dieser zurückbleibende Schleim ist ein guter Nährboden für Bakterien und Viren. Diese vermehren sich und es kommt zu einer chronischen Entzündung der Bronchien. Die äusserst feine Struktur der Bronchien und der ganzen Lunge wird durch die chronische Entzündung und die narbige Umwandlung empfindlich gestört. Ohne Therapie würde die Lunge durch den Sekretstau und die Entzündung der Bakterien allmählich zerstört. Dadurch kann die Lunge ihre Hauptaufgabe, den Sauerstoff der Luft ins Blut zu transportieren, nicht mehr wahrnehmen, und es entsteht ein Sauerstoffmangel.
Symptome in der Lunge
In unterschiedlichem Ausmass liegen folgende Symptome vor: Chronischer Husten, Aushusten von gelbem oder grünlichem Schleim, verstopfte Nase, Nasenpolypen, Kieferhöhlenentzündungen, eventuell
Blauverfärbung der Lippen und Fingerbeeren bei niedrigem Sauerstoffgehalt im Blut, eingeschränkte körperliche Leistungsfähigkeit.
Was kann und muss dagegen getan werden
- Regelmässige Inhalation von bronchienerweiternden Medikamenten (z.B. Ventolin und andere) damit derSchleim besser abgehustet werden kann.
- Inhalation von entzündungshemmenden Medikamenten (z.B. Pulmicort und andere), damit die chronischeEntzündung reduziert werden kann.
- Inhalation von schleimverflüssigenden Medikamenten (DNase: Pulmozyme), damit der zähe Schleim besser abgehustet werden kann, Inhalation von Antibiotika (TOBI, Colistin etc.), damit die Bakterien auch von der Bronchienseite angegriffen und reduziert werden können.
- Regelmässige, altersentsprechende Atemphysiotherapie zur Förderung des Schleimabhustens.
- Regelmässige sportliche Tätigkeit soweit möglich, ebenfalls zum besseren Abhusten des Schleims und zur Kräftigung der körperlichen Kondition.
- In regelmässigen, individuell aber unterschiedlichen Zeitabständen sogenannte Intensivkuren im Spital, bei welchen 14 Tage lang Antibiotika direkt in die Vene gegeben werden, intensive Atemphysiotherapie durchgeführt und kalorienreiche Nahrung angeboten wird
- Häufiger und länger als bei Kindern und Jugendlichen ohne CF werden auch Antibiotika als Tabletten oder Sirup eingesetzt. Wie bei der Gabe direkt in die Vene haben sie ebenfalls zum Ziel, die Anzahl der in der Lunge vorhandenen Bakterien zu reduzieren.
- Nach Bedarf wird Sauerstoff während der Nacht und/oder tagsüber sowie während der Physiotherapie eingesetzt
Diese Therapie ist zeitaufwändig und anspruchsvoll für die CF-Betroffenen und ihre Eltern. Das CF- Betreuerteam stellt die für den CF-Patienten optimale Therapie zusammen, erklärt die Notwendigkeit und die Wirkung, instruiert und unterstützt den Patienten und seine Eltern in der Durchführung.
Die Verdauungsorgane
Im Verdauungstrakt gibt es viele schleim- oder flüssigkeitsbildende Zellen. Auch dort ist der Schleim oder die produzierte Flüssigkeit zu zähflüssig. Besonders ungünstig ist dies im Falle des von der Bauchspeicheldrüse produzierten Verdauungssaftes. Der dickflüssige Bauchspeicheldrüsensaft verstopft die feinen Kanäle in der Bauchspeicheldrüse, und bald gelangt kein Saft mehr in den Darm. Dieser Saft wird aber gebraucht, um alle Nahrungsmittel, welche wir essen in winzig kleine Teilchen zu spalten; nur so können die Nahrungsmittel überhaupt im Darm aufgesaugt (resorbiert) werden. Wenn die Nahrungsmittel nicht mit diesem Verdauungssaft vermischt werden, können sie nicht im Dünndarm resorbiert werden, und es gehen alle Nährstoffe und Kalorien verloren. Zucker, Fett und Eiweiss gelangen in den Dickdarm. Dort werden diese Nahrungsbestandteile von den Bakterien der Darmflora abgebaut. Dies löst Blähungen und fettig glänzende Stühle oder Durchfall und Bauchschmerzen aus. Darum sind CF-Patienten meist trotz grosser Essensmengen sehr dünn und gedeihen nicht erwartungsgemäss. Der Verdauungssaft kann künstlich hergestellt werden und steht als Mikro-Kügelchen in Kapseln verpackt zur Verfügung (z.B. Creon, Panzytrat und andere).
Was man dagegen tun kann und muss
- Alle Nahrungsmittel, jegliches Essen auch und besonders der Schokoriegel zwischendurch muss von künstlichem Verdauungssaft (Creon, Panzytrat oder andere) begleitet sein. Von Beginn weg lernt der CF- Betroffene und seine Eltern, wie manche Kapsel er für die jeweilige Portion Essen braucht. Als Faustregel gilt: Je mehr Fett das Essen enthält, desto mehr künstlichen Verdauungssaft braucht es. Sind die Stühle fettig oder treten Bauchschmerzen auf, ist die Menge ungenügend und muss erhöht werden.
- Eine ganz genaue Vermischung der Nahrung mit dem künstlichen Verdauungssaft gelingt nicht, und es gehen immer Kalorien verloren. Deshalb und weil CF-Betroffene für die Atmung und die Bekämpfung der Bakterien in der Lunge mehr Energie brauchen, müssen sie mindesten 150 besser 200% des normalen Kalorienbedarfes zu sich nehmen. Kalorienreiche, fettreiche Nahrungsmittel sind daher ideal für CF- Betroffene
- Vitamine werden zusätzlich zugeführt. Die Vitamine A, D, E, und K sind sogenannte fettlösliche Vitamine. Sie brauchen ebenfalls künstlichen Verdauungssaft, damit sie im Darm aufgenommen werden können. Das heisst, die Vitamine müssen zum Essen und mit den Kapseln eingenommen werden. Dies entspricht nicht der gängigen Vorstellung einer gesunden Ernährung, für CF-Betroffene gilt jedoch die genügende Kalorienzufuhr als erste Priorität.
Bauchspeicheldrüse und Zuckerregulation
Die Bauchspeicheldrüse produziert nicht nur Verdauungssäfte sondern auch Insulin, ein Hormon, welches für die Zuckerregulation unverzichtbar ist. Bei fortschreitender Erkrankung (in der Regel im jugendlichen Alter) entwickelt einTeil der CF-Betroffenen eine Zuckerkrankheit oder Diabetes. Dies wird in den Kontrollen regelmässig gesucht und sobald als möglich behandelt.
Galle
Durch Rückstauung der Galle, auch sie ist bei CF-Betroffenen zu dickflüssig, kann eine mehr oder weniger starke Entzündung der Gallenwege und der Leber mit anschliessender Verhärtung entstehen. Wenn zuwenig Galle in den Darm fliesst, färben sich die Stühle beige-weiss und die Aufnahme von Fett aus der Nahrung ist erschwert. In diesem Falle kann ein Medikament, welches die Zusammensetzung der Galle verändert und sie flüssiger macht, eingesetzt werden. Bei manchen CF-Betroffenen ist die Verhärtung der Leber als Symptom im Vordergrund. In regelmässigen Kontrollen wird die Funktion der Leber überwacht.
„Mekoniumileus“
Bei ca. 10% der CF-Betroffenen ist der Stuhl, welcher während des vorgeburtlichen Lebens gebildet wird so zäh, dass er nicht wie erwartet innerhalb der ersten 48h abgesetzt werden kann. Im schlimmsten Fall verstopft er den Darm so sehr, dass dieser stark gebläht wird und sogar kleine Löcher entstehen können. Diese Situation ist hochverdächtig für das Vorliegen einer CF. Kinder mit dieser Symptomatik müssen nach der Geburt umgehend operiert und der betroffene Darmabschnitt entfernt werden.
Geschlechtsorgane
Aufgrund der genannten zähen Konsistenz der Samenflüssigkeit, aber auch wegen Anomalien der Samenleiter und Nebenhoden ist bei ca. 90% der männlichen CF-Patienten die Zeugungsfähigkeit eingeschränkt. Dank neueren urologischen Methoden können diese Probleme aber überbrückt werden. Bei Frauen erschwert eine starke Verschleimung mit Pfropfenbildung in den Eileitern die Entstehung einer Schwangerschaft beträchtlich. Eine Schwangerschaft ist aber nicht unmöglich.
CF-Betreuungsteam
Die Instruktion und Anpassung der komplexen Therapie und die gute Verlaufsbeobachtung der Krankheit sowie das Einleiten der nötigen medizinischen und sozialen Massnahmen erfordert eine optimale
Zusammenarbeit zwischen den Eltern, den Ärzten im Spital und in der Praxis sowie den Physiotherapeuten, Sozialarbeitern und den Ernährungsberaterinnen. Dies wird durch regelmässige Kontrollen im Zentrumsspital und eine gute Zusammenarbeit aller Beteiligten erreicht.
Medizinische Massnahmen – Behandlung
- CF-Betroffene müssen viel essen und während den Mahlzeiten Verdauungsenzyme einnehmen.
- Durch mehrmalige tägliche Inhalationen und Atemtherapien werden die Auswirkungen der CF in der Lunge gelindert. Diese physiotherapeutischen Massnahmen wirken auch vorbeugend, denn sie begünstigen gute Verhältnisse im Atmungstrakt.
- Zur Unterstützung der Therapien eignen sich Gymnastik zur Erhaltung der Beweglichkeit und eine dem
- Gesundheitszustand angepasste sportliche Betätigung.
- Zusätzlich dazu ist jedes Jahr ein- oder mehrmals eine intravenöse Antibiotika-Kur (i.v.-Kur) im Spital oder zu Hause notwendig.
- Durch eine Organtransplantation ist es heute möglich, dem Leben eines CF-Betroffenen viele Jahre hinzuzufügen.
- Intensiv-Therapie-Lager für Erwachsene fördern die sozialen und kreativen Fähigkeiten und die Krankheitsbewältigung des Einzelnen. Die Teilnehmer erlernen neue Atemtechniken zur Reinigung der Lunge und erleben die Gemeinschaft innerhalb einer Gruppe von Gleichaltrigen mit derselben Erkrankung.
- Die psychologische und psychosoziale Betreuung der CF-Betroffenen nimmt zusehends einen hohen Stellenwert ein.
CF ALS SELTENE KRANKHEIT
[12.2012] Spricht man von Cystischer Fibrose, spricht auch immer davon, dass es sich um eine sogenannt seltene Krankheit handelt. Doch wieso ist diese Betonung der Seltenheit so wichtig, was unterscheidet seltene Krankheiten von anderen und warum ist diese Betrachtungsweise so relevant?
Seltene Krankheiten im Allgemeinen
Seltene Erkrankungen betreffen eine beschränkte Zahl von Menschen und bringen wegen ihrer Seltenheit besondere Probleme mit sich. In Europa ist eine Krankheit selten, wenn sie weniger als einen von 2000 Einwohnern betrifft. Bei CF in der Schweiz liegt diese Quote bei etwa einem Fall auf 2500 Geburten.
Es gibt Tausende seltener Krankheiten. Derzeit sind zwischen 6000 und 8000 bekannt, und in der Fachliteratur werden regelmässig neue beschrieben. 80 % der seltenen Krankheiten sind genetischer Natur (wie auch die Cystische Fibrose). Zu den übrigen 20 % gehören extrem seltene Infektionskrankheiten, Autoimmunkrankheiten und seltene Krebsformen. Sämtliche Bereiche der Medizin sind betroffen. Für eine Vielzahl von Erkrankungen kennt man den Grund bis heute nicht.
Es gibt zwar keine umfassenden Studien über die Häufigkeit seltener Erkrankungen. Es dürften aber 6 % bis 8 % der globalen Bevölkerung oder 1 Mensch von 17 betroffen sein. Insgesamt leiden rund 30 Millionen Europäer und 27 Millionen Nordamerikaner an einer seltenen Krankheit. In der Schweiz gibt es praktisch keine epidemiologischen Daten über seltene Erkrankungen. Hochrechnungen aufgrund europäischer Statistiken lassen aber auf eine Zahl von 500 000 Schweizern schliessen. Trotz unterschiedlichster Eigenschaften haben diese Krankheitsbilder auch vieles gemeinsam. Sehr oft sind sie schwer, verlaufen chronisch und können lebensbedrohend sein. Diese Beeinträchtigung führt oft zu eingeschränkter Selbständigkeit der Betroffenen, schmälert deren Lebensqualität und mündet in eine Behinderung. Die gemeinsamen Herausforderungen, auf die alle Menschen mit einer seltenen Krankheit treffen, sind in etwa die folgenden:
- Es erfolgt keine korrekte Diagnosestellung
- Die Diagnose erfolgt oft (zu) spät oder nach langen Verzögerungen
- Es gibt nur wenige und unzureichende Informationen über die Krankheit
- Die Krankheit wird auf wissenschaftlicher Ebene nicht genügend verstanden Die Krankheit hat schwerwiegende soziale Probleme für die Betroffenen zur Folge
- Es fehlt eine angepasste und spezialisierte medizinische Behandlung
- Ungleichbehandlung und Schwierigkeiten beim Zugang zu Therapien und Behandlung
- Selbst Ärzte und das medizinische Fachpersonal kennen die seltenen Krankheiten meist nur ungenügend. Oft ist auch von «Orphan»-Erkrankungen die Rede (also «vernachlässigte Weisenkrankheiten»). Hier fehlt es an medizinischer Forschung und an spezifischen Behandlungen, wenn es überhaupt welche gibt.
Im Bereich der Medikamenten- und Wirkstoffentwicklung interessierte sich die Pharmaindustrie bis vor Kurzem nur in begrenztem Mass für die Entwicklung von Orphan Drugs. Grund ist die tiefe Rentabilität wegen eines im Vergleich mit häufiger vorkommenden Erkrankungen extrem kleinen Marktes. Damit wäre auch erklärt, warum verschiedenste nicht gewinnorientierte Organisationen wie zum Beispiel die Aktion Telethon Schweiz oder auch die CFCH nach wie vor Mittel für die Erforschung dieser Krankheiten beschaffen.
Die Erforschung seltener Erkrankungen ist mittlerweile aber interessant, denn heute weiss man: Seltene Erkrankungen dienen oft als Modell für häufiger vorkommende Krankheitsbilder, weshalb sich die Forschungsergebnisse später oft auszahlen. Für eine optimale Forschungsarbeit bedarf es einer engen Zusammenarbeit der Experten weltweit und des Einbezugs einer möglichst hohen Zahl von Patienten. Mit einer guten Koordination lässt sich Expertenwissen aus verschiedenen Berufsgattungen austauschen.
CF als seltene Krankheit
Zusammenfassend kann also festgehalten werden, dass Menschen mit seltenen Krankheiten auf ihrem Weg zur Diagnose, bei der Suche nach Informationen und kompetenten Fachkräften alle auf ähnliche Schwierigkeiten stossen. Ein Zugang zu angepassten Therapien, eine umfassende soziale und medizinische Betreuung, die Koordination der Spital- und der ambulanten Pflege, Selbstständigkeit, soziale und berufliche Eingliederung sind keine Selbstverständlichkeit. Vom psychischen Leidensdruck infolge der Isolation und der mangelnden Hoffnung auf Heilung gar nicht zu reden.
Aber treffen diese Aussagen heute auch noch vollumfänglich auf Menschen mit Cystischer Fibrose zu? Dies ist sicherlich nicht der Fall. Auch wenn hier nichts schöngeredet werden soll, ist doch festzuhalten, dass in zahlreichen angloamerikanischen und westeuropäischen Ländern Menschen mit CF weitaus besser gestellt sind als andere Betroffene von seltenen Krankheiten. In der Schweiz verfügen wir mittlerweile über ein CF-Screening an Neugeborenen, was eine rasche Diagnosestellung ermöglicht, und seit Jahrzehnten über hochspezialisierte Behandlungszentren in verschiedenen Regionen der Schweiz. Zudem können sich Eltern und CF-Betroffene nicht zuletzt über die CFCH miteinander vernetzen, um sich über den Umgang mit der Krankheit auszutauschen. Womit keinesfalls gesagt sein soll, dass das Leben mit CF heute ein einfaches Schicksal wäre, es bestehen nach wie vor Ungleichbehandlungen und Schwierigkeiten.
Aber bei der Cystischen Fibrose ist es seit der ersten korrekten medizinischen Beschreibung der Krankheit 1938 gelungen, dank engagierten Personen Schritt ein die richtige Richtung zu tun. So wurde in den 1950er- und 1960er-Jahren in den USA und Europa die ersten spezialisierten CF-Zentren und ersten CF-Organisationen gegründet. Nach und nach konnten so die typischen Herausforderungen, welche oben beschrieben sind und auf alle seltenen Krankheiten zutreffen, zumindest etwas entschärft, wenn nicht ganz beseitigt werden.
Auf dieser Basis ist es bis heute gelungen, Patienten, das betreuende medizinische Personal und Forschende weltweit zu vernetzen. Dieser Umstand ist bei einer seltenen Krankheit zentral. Je mehr «Fälle» der Krankheit weltweit beschrieben und behandelt werden, umso grösser sind die Chancen auf wissenschaftlichen Fortschritt und bessere Behandlung. Die folgende Abbildung beschreibt eindrücklich, welchen Einfluss neue wissenschaftliche Erkenntnisse auf den Verlauf des Durchschnittsalters bei CF haben, dass sich in den vergangenen Jahrzehnten mehr als verdreifacht hat.
Die Notwendigkeit der internationalen Kooperation bei seltenen Krankheiten ist auch für die CFCH unbestritten. Dies ist auch einer der Gründe weshalb unsere Organisation dieses Frühjahr dem Clinical Trial Network (europäisches Netzwerk zur Förderung der klinischen Studien) seine Unterstützung zugesagt hat. Im Rahmen des europäischen Austausches unter den seltenen Krankheiten gelten die nationalen CF-Gesellschaften und deren europäische Dachorganisation CF-Europe als beispielhaft für eine grenz übergreifende Vernetzung. Dasselbe gilt für die nationalen CF-Ärzteorganisationen und deren europäische Dachorganisation European Cystic Fibrosis Society (ECFS).
Bleibende Herausforderungen
Trotz aller positiven Entwicklungen der letzen Jahrzehnte bleiben natürlich Herausforderungen bestehen. CF ist nach wie vor nicht heilbar.
Daher engagiert sich die CFCH neu auch in der Organisation ProRaris – Allianz Seltener Krankheiten – Schweiz für die Anliegen von Menschen mit seltenen Erkrankungen. Der gut vernetzten Organisation ist es gelungen, die Eidgenossenschaft auf unsere gemeinsamen Anliegen hinzuweisen.
Quelle: CFCH